

四、凝血及抗凝血因子缺乏症

凝血是一复杂的生理过程,包括多种凝血因子及抗凝血因子参与。遗传性凝血障碍包括低凝和高凝两种情况。前者是由于基因突变致凝血因子活性降低,后者是由于抗凝血因子活性缺乏。
1.凝血因子缺乏症
除Ca2+与凝血活素外,所有凝血因子都有遗传性缺乏的报告,其中血友病,特别是甲型血友病较为多见。现主要就血友病及与其有关的血管性假血友病加以介绍。
血友病(hemophilia)除甲、乙、丙三型外,加上后来又发现的一种vWF因子(vonwillibrand factor)缺乏的血管性假血友病,构成血友病的四种类型。1986-1989年全国24省、市,37个地区作血友病患病率的调查,调查人数16866 654人,总患病率2.73/10万(男性5.21/10万,女性0.06/10万),与欧美相比较低,四型的构成比是:甲型79.8%,乙型14.1%,丙型2.8%,血管性假血友病3.3%。
(1)甲型血友病:甲型血友病(hemophilia A)又名抗血友病球蛋白(antihemophilicglobulin,AHG)缺乏症或第Ⅷ因子缺乏症。主要表现为出血倾向,其出血特点为:①缓慢持续渗血;②多发生于轻微创伤之后;③出血部位广泛,常反复发生,可形成血肿,并节变形,死因多为颅内出血。
现知,Ⅷ因子由三种成分组成:①FⅧ:C(AHG);②FⅧ:Ag(Ⅷ因子相关抗原);③vWF因子。甲型血友病为AHG遗传性缺乏所致。
本病为X连锁隐性遗传,基因定位于Xq28,基因跨度超过186kb,由26个外显子(占9kb)及25个内含子(占177kb)组成,编码2351个氨基酸,已发现缺失型(包括错义、无义及移码突变等)共46种以上。杂合子的鉴定对开展遗传咨询很重要。过去多通过测定血浆AHG水平或用ⅧR:Ag/AHG比值来检出杂合子,现已能采取分子遗传学手段,特别是已成功地应用DNA印迹杂交、PCR技术等于产前诊断,这对防止重型患儿出生,十分有效。
治疗可使用各种AHG制剂,但需长期使用,正在研究的基因治疗将会是本病的根治方法。
(2)乙型血友病:乙型血友病(hemophilia B)又名血浆凝血活酶成分(PTC)缺乏症或第Ⅸ因子缺乏症。此型临床表现酷似甲型,但发病率较低,遗传方式亦为Ⅹ连锁隐性遗传,由于杂合子Ⅸ因子活性仅为正常1/3,某些杂合子可出现症状,故女性病人较甲型多见。
人类第Ⅸ因子基因已定位Xq27.1,基因总长度为34kb左右,由8个外显子组成。已鉴定出的突变有100种之多(部分缺失及全缺失者30种,其余为各种类型点突变)。我国王宁波等报告了重庆发现的5种点突变(外显子2和5)。上海曾溢滔等发现1例1-3内含子5kb片段缺失。在这些突变中发现几例启动子突变,临床表现为儿童期有严重出血倾向,但到青春期后自发出血减轻。后证明,雄性类固醇可诱导启动子产生Ⅸ因子,这也表明临床症状轻重与突变性质可以有一定关系。
本型亦多采用输血浆或浓缩血浆制剂治疗,将第Ⅸ因子活性提高到25%以上即有疗效。产前诊断是防止本病患儿出生的有效方法。近年,我国薜京伦等在Ⅸ因子缺乏症的基因治疗方面取得了进展,有望在临床取得长期稳定疗效。
(3)丙型血友病:丙型血友病(hemophilia C)又名血浆凝血活酶前质(PTA)缺乏症(plasma thromboplastic antecedent deficiency)或第Ⅸ因子缺乏症。此型症状较甲、乙型轻。本病种族倾向明显,多见于土耳其南部犹太人后裔。遗传方式属染色体隐性遗传。现知,基因定位于15q11,基因长度为23kb,由15个外显子组成,编码625个氨基酸,但仅第11-15外显子编码羧基端具有凝血功能为Ⅺ因子主要成分。已发现3种点突变。本病纯合子的Ⅺ因子活性在20%以下,杂合子为30%-65%。多数严重缺乏者用小剂量正常或浓缩血浆治疗即显效。
(4)血管性假血友病:血管性假血友病(von Willebrand disease)是一种较多见的与第Ⅷ因子有关的遗传性凝血障碍。与本病有关的von Willebrand因子(v WF)是一大分子量的糖蛋白。基因定位于12pter-p12,vWF基因长度为180kb,有52个外显子,mRNA总长度为9kb左右编码2813个氨基酸。vWF蛋白由血管内皮细胞及巨噬细胞分泌,它在血中不仅作为Ⅷ因子载体,而且可增强Ⅷ稳定性,故vWF因子缺乏往往伴有Ⅷ因子活性降低。此外,血小板α颗粒中含vWF,故也参与血小板聚集,在凝血中发挥作用。本病患者有明显的出血倾向。血中AGH活性降低,但不如甲型血友病严重,从基因水平已发现20多种突变型。
由于对本病基因有足够的了解,已可通过RFLP连锁分析或PCR法对本病进行产前诊断。
此外,由于遗传性血小板缺乏或功能障碍和纤维蛋白原的遗传性缺陷都可造成低凝状态,在此不多赘述。
2.抗凝血因子缺乏症
(1)遗传性抗凝血酶Ⅲ缺乏症:抗凝血酶Ⅲ(antithrombinⅢ,ATⅢ)对凝血酶Xa有抑制作用,肝素能加速其对凝血酶的抑制。其次,ATⅢ还有抑制Ⅸ、Ⅺ及Ⅻ的功能。
遗传性抗凝血酶Ⅲ缺乏症(hereditary antithrombin Ⅲ deficiency)的临床表现为容易发生血栓形成(主要部位为髂静脉)及肺体塞。
本病为常染色体显性遗传。发病率在不同种族有显著差异。欧美白种人中可高1:2000-5000。我国也已有个例报告。现知,ATⅢ基因定位1q23,基因长16kb,由7个外显子组成,编码432个氨基酸,至少已发现20种以上的突变类型,表现出不同的功能缺陷。
(2)遗传性蛋白C系统异常:遗传性蛋白C系统由蛋白C(PC)、蛋白S(PS)、血栓调节蛋白(thrombomdorlin,TM)及活性蛋白C抑制物(APC1)组成,它们之间的关系见图4-19。活化的蛋白C有抑制Va、Ⅻa及促进纤维蛋白溶解的作用。蛋白C系统缺乏症有3种:
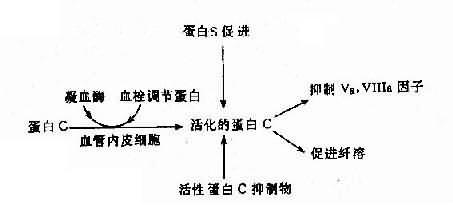
图4-19 蛋白C、蛋白S及活性蛋白C抑制物的相互关系
1)蛋白C缺乏症:主要症状是静脉血栓形成,常见于深部静脉。本病发生率估计为1:16000,呈常染色体显性(或不完全显性)遗传,纯合子严重,表现为出血性皮肤坏死、弥漫性血管内凝血(DIC)和血栓。杂合子多数在青壮年发病。蛋白C基因定位2号染色体,基因长12kb,由8个外显子组成,已鉴定出若干种缺失型及点突变病例,也有涉及mRAN加工缺陷者。
2)蛋白S缺乏症:蛋白S的作用是促进活化蛋白C(APC)结合于磷脂,加速APC灭活Va因子。本病亦属常染色体显性(或不完全显性)遗传。基因定位在3号染色体,其总长度为45kb。上海也已报告2例PS缺乏症。
3)先天性活化蛋白C抑制缺乏症(congenitaldeficiency of activated protein Cinhibitor)。
其它还有遗传性纤溶系统异常,包括①先天性异常纤溶酶原血症;②先天性纤溶酶原激活物释放异常;③遗传性纤溶酶原激活抑制物增多症;④血块异常所致先天性纤溶减弱等,均表现出高凝状态的各种症状。
- 凝血及抗凝血因子缺乏症《医学遗传学基础》
- 凝血和纤溶的变化《病理生理学》
- 凝血酶活性测定法《中国生物制品规程》
- 凝雪汤《备急千金要方》
- 凝血酶原的激活《生物化学与分子生物学》
- 凝形殊禀章《妇人大全良方》
- 凝血酶原激活物的生成《生物化学与分子生物学》
- 凝唾汤《备急千金要方》
- 凝血酶原时间《常用化验值及意义》
- 凝水石《本草图经》
- 凝血酶原消耗试验《常用化验值及意义》
- 凝水石《药性切用》
- 凝血酶原消耗试验《常用化验值及意义》
- 凝水石《本草撮要》
- 凝血时间《常用化验值及意义》
- 凝水石《本草分经》
- 凝血时间测定《临床基础检验学》
- 凝水石《名医别录》
- 凝血系统在ARDS发病中的作用《病理生理学》
- 凝水石《增广和剂局方药性总论》
- 凝血作用的调节《生物化学与分子生物学》
- 凝水石《得配本草》
- 凝壅聚结辨《痧胀玉衡》
- 凝水石《本草从新》
- 凝脂翳《中医名词词典》
- 凝水石《本经逢原》
- 凝脂翳《中医词典》
- 凝水石《证类本草》
- 凝脂翳《中医词典》
- 凝水石《医学入门》
- 凝脂翳《中医眼科学》